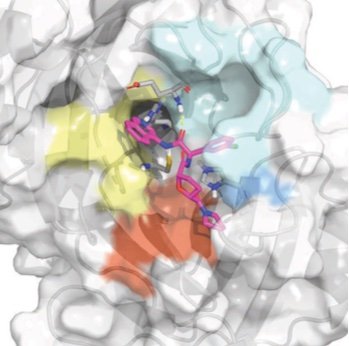
Prospective evaluation of structure-based simulations reveal their ability to predict the impact of kinase mutations on inhibitor binding
/
Sukrit Singh, Vytautas Gapsys, Matteo Aldeghi, David Schaller, Aziz M Rangwala, Jessica B White, Joseph P Bluck, Jenke Scheen, William G Glass, Jiaye Guo, Sikander Hayat, Bert L de Groot, Andrea Volkamer, Clara D Christ, Markus A Seeliger, John D Chodera.
[bioRxiv]
We show that alchemical free energy calculations have the potential to prospectively predict the impact of clinical kinase mutations on targeted kinase inhibitor binding.