Identifying ligand binding sites and poses using GPU-accelerated Hamiltonian replica exchange molecular dynamics
/
Kai Wang K, John D. Chodera, Yanzhi Yang, and Michael R. Shirts.
J. Comput. Aid. Mol. Des. 27:989, 2013. [DOI] [PDF]
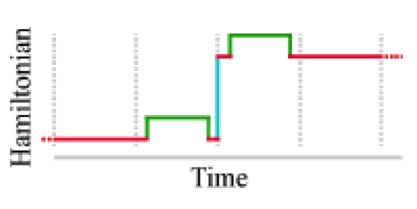
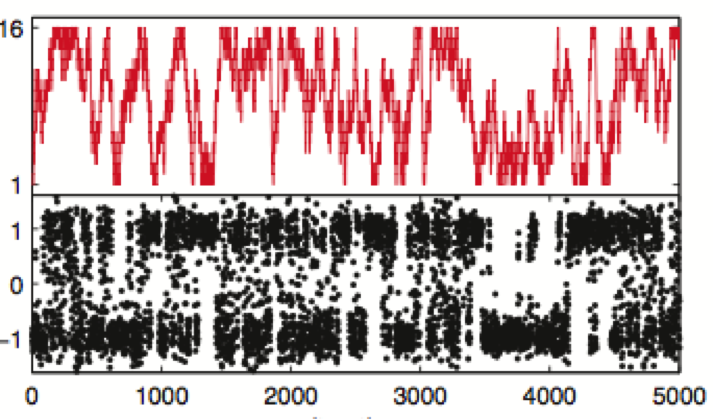
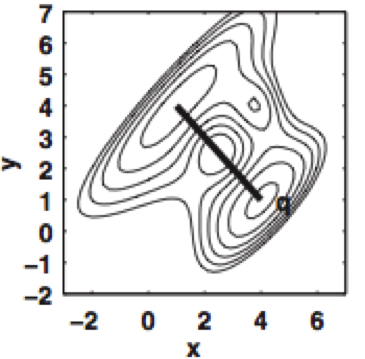
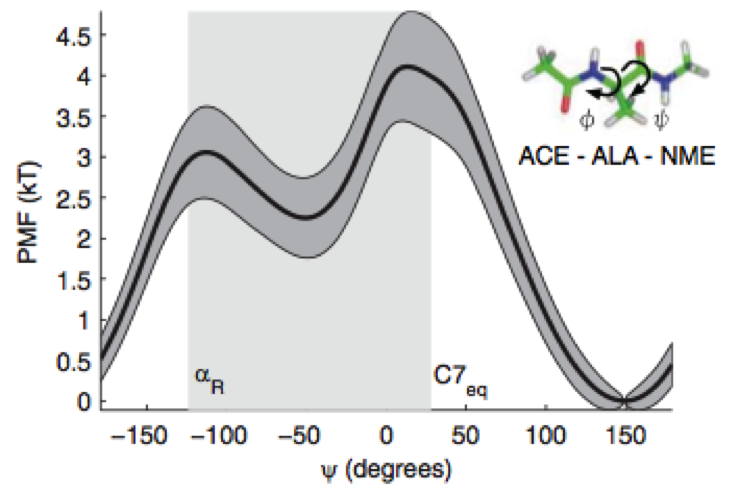
We show how bound ligand poses can be identified even when the location of the binding sites are unknown using the machinery of alchemical modern free energy calculations on graphics processors.